![]()
Experiencia Piloto de Evaluación Continuada para la Asignatura de “Aplicaciones de la Química Cuántica” de 3º de Ciencias Químicas.
Curso 2005/06
Tercer Ejercicio: Para poder entregar este tercer ejercicio será obligatorio asistir a la clase práctica. Mira aqui para saber las fechas de realización de la parte práctica. El último día para entregar el ejercicio es el jueves 19 de enero de 2006.
· Obtener el espectro de emisión de una llama y conocer qué moléculas (intermediatos) se forman.
·
A partir del espaciado vibracional
de las cabezas de banda del sistema Swan correspondiente a las
transiciones
d3Πg![]() a3Πu
de la molécula de
C2 obtener las constantes vibracionales
a3Πu
de la molécula de
C2 obtener las constantes vibracionales
![]() ,
,![]() ,... y
,... y
![]() ,
,![]() , ...
de los estados electrónicos implicados en esta banda
electrónica.
, ...
de los estados electrónicos implicados en esta banda
electrónica.
· Obtener las energías de disociación D0 de los estados d3Πg y a3Πu de la molécula de C2 mediante el método de extrapolación lineal de Birge-Sponer.
·
Comparar las intensidades
experimentales, de algunas bandas vibracionales del sistema
d3Πg![]() a3Πu
de la molécula de
C2, con las intensidades teóricas.
a3Πu
de la molécula de
C2, con las intensidades teóricas.
·
A partir del espaciado rotacional de
la rama P, de la banda ![]() ’=0-
’=0-
![]() ”=0 del
sistema
B2Σ-
”=0 del
sistema
B2Σ-![]() X2Π de la molécula de CH, se obtendrán
las correspondientes constantes rotacionales
Bv’=0 y Bv”=0.
Estimar también la temperatura de la llama mediante medidas
espectroscópicas.
X2Π de la molécula de CH, se obtendrán
las correspondientes constantes rotacionales
Bv’=0 y Bv”=0.
Estimar también la temperatura de la llama mediante medidas
espectroscópicas.
Cualquier reacción de combustión (por ejemplo: propano o butano con oxígeno) se produce a través de un gran número de procesos elementales. En las llamas se forman diferentes moléculas diatómicas o radicales, tales como C2, OH y CH en estados electrónicos excitados. Los espectros de estas moléculas se deben a transiciones electrónicas (UV-Visible) entre distintos estados vibracionales y rotacionales.

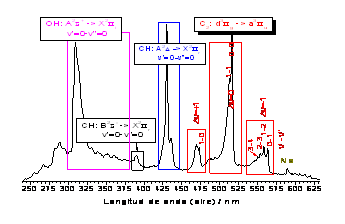
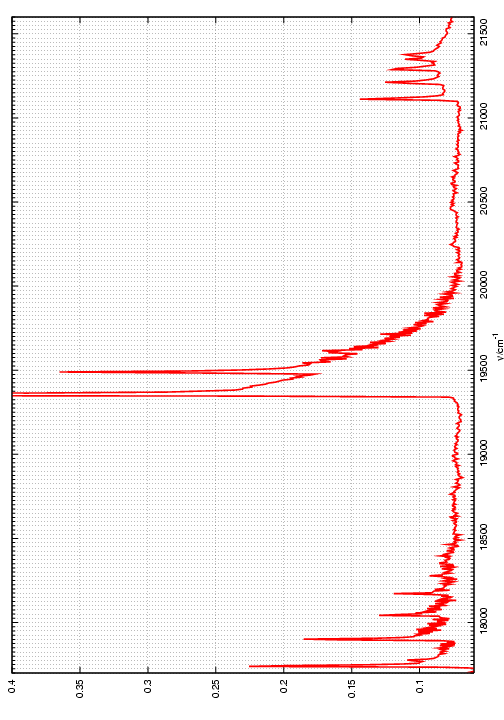
Figuras 1 y 2: Espectros de emisión en función de la longitud de onda y del número de ondas.
En la figura superior derecha
(Figura 1) se indica la zona donde aparecen las bandas
electrónicas de las diferentes moléculas diatómicas (radicales)
formadas en la llama: C2:
d3Πg->
a3Πu (banda Swan) con Δ
Δ![]() =-1, 0, 1;
CH: A2Δ->
X2Π (
=-1, 0, 1;
CH: A2Δ->
X2Π (![]() ’=0-
’=0-![]() ”=0)
y
B2Σ-
->X2Π (
”=0)
y
B2Σ-
->X2Π (![]() ’=0-
’=0-![]() ”=0)
y OH:
A2Σ+->
X2Π (
”=0)
y OH:
A2Σ+->
X2Π (![]() ’=0-
’=0-![]() ”=0).
En la figura superior izquierda (Figura 2) se muestra
el espectro de la llama obtenido con mayor resolución, donde
ahora la escala en el eje de abcisas corresponde al número de
onda (en cm-1) en vacío.
”=0).
En la figura superior izquierda (Figura 2) se muestra
el espectro de la llama obtenido con mayor resolución, donde
ahora la escala en el eje de abcisas corresponde al número de
onda (en cm-1) en vacío.
Las líneas espectrales de muchas
moléculas no hidruros como C2 se agrupan en
secuencias de banda que corresponden a transiciones con
igual Δ![]() =
=![]() ’-
’-![]() ”.
Esto sucede cuando las constantes vibracionales de los estados
electrónicos implicados no son muy diferentes (el espaciado
vibracional es similar). Como resultado las transiciones con
igual Δ
”.
Esto sucede cuando las constantes vibracionales de los estados
electrónicos implicados no son muy diferentes (el espaciado
vibracional es similar). Como resultado las transiciones con
igual Δ![]() aparecen juntas en el espectro.
La energía vibracional, en términos de energía
en
aparecen juntas en el espectro.
La energía vibracional, en términos de energía
en
cm-1 , se puede expresar como un desarrollo en serie:
![]() (1)
(1)
donde ![]() ,
, ![]() ,…, son
las constantes vibracionales (en cm-1). Para
transiciones de una misma banda electrónica, el espaciado
entre niveles vibracionales adyacentes viene dado
por
,…, son
las constantes vibracionales (en cm-1). Para
transiciones de una misma banda electrónica, el espaciado
entre niveles vibracionales adyacentes viene dado
por
![]() (2)
(2)
Si se consideran bandas con ![]() ’
constante (progresiones
’
constante (progresiones ![]() ’),
a partir del espaciado entre bandas adyacentes donde cambia
’),
a partir del espaciado entre bandas adyacentes donde cambia
![]() ”,
se pueden obtener las constantes vibracionales del estado
fundamental
”,
se pueden obtener las constantes vibracionales del estado
fundamental ![]() ,
,![]() , etc. Si se
consideran bandas con
, etc. Si se
consideran bandas con ![]() ”
constante (progresiones
”
constante (progresiones ![]() ”),
a partir del espaciado entre bandas adyacentes donde cambia
”),
a partir del espaciado entre bandas adyacentes donde cambia
![]() ’,
se pueden obtener las constantes vibracionales del estado
excitado
’,
se pueden obtener las constantes vibracionales del estado
excitado ![]() ,
,![]() ,etc. Para estos
cálculos hay que utilizar la ecuación (2), aunque con el fin
de simplificar el análisis, nos quedaremos únicamente con los
dos primeros términos. En la Figura 3 se indica la
asignación vibracional (cabezas de banda) que tomaremos como
orígenes de banda debidos al sistema de bandas
d3Πg
,etc. Para estos
cálculos hay que utilizar la ecuación (2), aunque con el fin
de simplificar el análisis, nos quedaremos únicamente con los
dos primeros términos. En la Figura 3 se indica la
asignación vibracional (cabezas de banda) que tomaremos como
orígenes de banda debidos al sistema de bandas
d3Πg![]() a3Πu
de la molécula de
C2.
a3Πu
de la molécula de
C2.
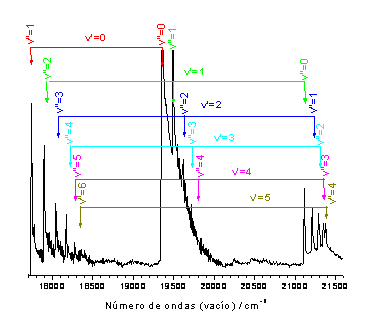
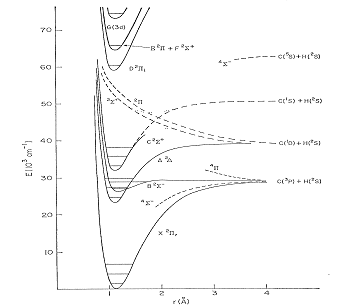
Figura 3: Asignación de las cabezas de banda del sistema d3Πg -> a3Πu del C2. Figura 4: Curvas de energía potencial del CH.
Ejercicio 1:
Teniendo en cuenta esta
asignación, a partir de los orígenes de banda medidos (en
cm-1) en el espectro dado, completar la
tabla de Deslandres indicada a continuación para el
sistema d
3Πg![]() a 3Πu
de la molécula de
C2. Para rellenar la Tabla 1 con los
datos correspondientes a las transiciones con
Δ
a 3Πu
de la molécula de
C2. Para rellenar la Tabla 1 con los
datos correspondientes a las transiciones con
Δ![]() =-1, 0, 1, utilizad los datos del fichero
adjunto, representado en la Figura 3. A partir de las
primeras diferencias de las cabezas de banda obtener los
valores promedios de las diferencias <Δνv”,v”+1>
y
<Δνv’,v’+1>
. Representar estos valores promedio en función del número
cuántico vibracional (
=-1, 0, 1, utilizad los datos del fichero
adjunto, representado en la Figura 3. A partir de las
primeras diferencias de las cabezas de banda obtener los
valores promedios de las diferencias <Δνv”,v”+1>
y
<Δνv’,v’+1>
. Representar estos valores promedio en función del número
cuántico vibracional (![]() ” o
” o
![]() ’) y
realizar un ajuste a la
ecuación (2) con sólo dos constates vibracionales, con el fin
de obtener
’) y
realizar un ajuste a la
ecuación (2) con sólo dos constates vibracionales, con el fin
de obtener ![]() y
y
![]() para los estados
electrónicos d3Πg y
a3Πu de la molécula de
C2 y comparar con los valores
tabulados. Representar en la misma figura el ajuste
realizado.
para los estados
electrónicos d3Πg y
a3Πu de la molécula de
C2 y comparar con los valores
tabulados. Representar en la misma figura el ajuste
realizado.
Tabla 1:
tabla de Deslandres para las
bandas vibracionales con Δ![]() =-1, 0, 1, correspondiente a la transición
electrónica d
3Πg
=-1, 0, 1, correspondiente a la transición
electrónica d
3Πg![]() a 3Πu
de la molécula de
C2.
a 3Πu
de la molécula de
C2.
|
|
v’=0 |
|
v’=1 |
|
v’=2 |
|
v’=3 |
|
v’=4 |
|
v’=5 |
<Δνv”,v”+1> |
|
v”=0 |
19352.3 |
1759.1 |
21111.4 |
|
|
|
|
|
|
|
|
|
|
|
1613.0 |
|
|
|
|
|
|
|
|
|
|
|
|
v”=1 |
17739.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
v”=2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
v”=3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
v”=4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
v”=5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
<Δνv’,v’+1> |
|
|
|
|
|
|
|
|
|
|
|
|
Cuando se conocen las frecuencias
vibracionales de diferentes transiciones entre niveles
vibracionales adyacentes ![]() =0↔
=0↔![]() =1,
=1, ![]() =1↔
=1↔![]() =2,
=2, ![]() =2↔
=2↔![]() =3,
... se puede utilizar una técnica gráfica conocida como
extrapolación de Birge-Sponer para obtener una energía de
disociación D0, referida al nivel vibracional más
bajo, de un estado electrónico. El método gráfico de Birge-Sponer
se basa en que la suma de los sucesivos intervalos
ΔGv+1/2 (ecuación 2) desde el nivel de
punto cero de energía hasta el límite de disociación es la energía
de disociación:
=3,
... se puede utilizar una técnica gráfica conocida como
extrapolación de Birge-Sponer para obtener una energía de
disociación D0, referida al nivel vibracional más
bajo, de un estado electrónico. El método gráfico de Birge-Sponer
se basa en que la suma de los sucesivos intervalos
ΔGv+1/2 (ecuación 2) desde el nivel de
punto cero de energía hasta el límite de disociación es la energía
de disociación:
![]() (3)
(3)
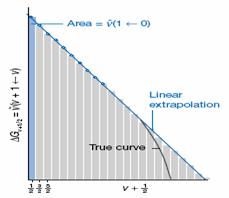 Si se representa
ΔGv+1/2 frente a
Si se representa
ΔGv+1/2 frente a
![]() +1/2,
el área bajo la curva es dicha suma y dará la energía de
disociación D0. Los términos sucesivos
ΔGv+1/2 decrecen casi linealmente
con
+1/2,
el área bajo la curva es dicha suma y dará la energía de
disociación D0. Los términos sucesivos
ΔGv+1/2 decrecen casi linealmente
con ![]() +1/2 (Figura 5) con lo que si se extrapola
linealmente dicha gráfica se obtiene un valor aproximado de la
energía de disociación D0 , superior al valor
real.
+1/2 (Figura 5) con lo que si se extrapola
linealmente dicha gráfica se obtiene un valor aproximado de la
energía de disociación D0 , superior al valor
real.
Figura 5: Extrapolación de Birge-Sponer. El área bajo la curva es igual a la energía de disociación D0.
Ejercicio 2:
Teniendo en cuenta los datos de la tabla de Deslandres del
Ejercicio 1 (Tabla 1) representar ΔGv’+1/2frente a ![]() ’+1/2 para obtener por extrapolación lineal la energía
de disociación del estado superior
d3Πg de la molécula de
C2. Comparar con el valor calculado
como
’+1/2 para obtener por extrapolación lineal la energía
de disociación del estado superior
d3Πg de la molécula de
C2. Comparar con el valor calculado
como
![]() (4)
(4)
donde para el
cálculo de la energía de disociación De referida
al mínimo de la curva de energía potencial se ha utilizado la
relación de Kratzer. Obtener G(0) con las constantes dadas
en la Tabla 3. Representar
ΔG v”+1/2 frente a
![]() ”+1/2
para obtener por extrapolación lineal la energía de disociación del
estado inferior a3Πu de la
molécula de C2. Comparar con el valor calculado
utilizando la ecuación (4).
”+1/2
para obtener por extrapolación lineal la energía de disociación del
estado inferior a3Πu de la
molécula de C2. Comparar con el valor calculado
utilizando la ecuación (4).
La intensidad de una banda
vibracional ![]() ’-
’- ![]() ” en emisión se puede aproximar
como
” en emisión se puede aproximar
como
![]() (5)
(5)
donde C es una constante, Nv’ es la población del estado excitado desde donde se produce la emisión y
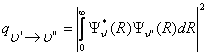 (6)
(6)
es el factor de Franck-Condon. Para transiciones
electrónicas, las funciones de onda vibracionales
Ψv(R) corresponden a estados electrónicos
diferentes. Cuanto mayor es el solapamiento de las funciones de
onda mayor es la intensidad de la banda. Como ejemplo, en la
Figura 6 se muestran las curvas de energía potencial y
algunas funciones densidad de probabilidad
![]() para los estados electrónicos
B1Πu y
X1Σ+ de la molécula de
Na2.
para los estados electrónicos
B1Πu y
X1Σ+ de la molécula de
Na2.
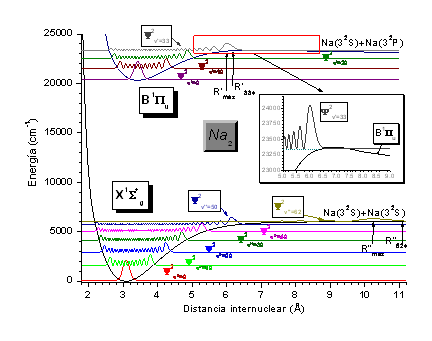
Figura 6: Curvas de energía potencial y funciones densidad de probabilidad de algunos estados de la molécula de Na2.
A presión atmosférica se producen un gran número de colisiones y las moléculas en estados electrónicos excitados se redistribuyen siguiendo una distribución tipo Boltzmann:
![]() (7)
(7)
donde N0 es la población del
nivel vibracional más bajo (que se puede tomar como 1),
G(![]() )-G(0) es la diferencia de energía vibracional entre
los estados con número cuantico vibracional
)-G(0) es la diferencia de energía vibracional entre
los estados con número cuantico vibracional
![]() y el nivel vibracional más bajo
(
y el nivel vibracional más bajo
(![]() =0) y T es la temperatura
absoluta.
=0) y T es la temperatura
absoluta.
Tabla 2:
Factores de Franck-Condon
(Ecuación 6) para algunas bandas vibracionales del espectro
electrónico
d3Πg![]() a3Πu
de la molécula de
C2.
a3Πu
de la molécula de
C2.
|
v” |
v’=0 |
v’=1 |
v’=2 |
v’=3 |
v’=4 |
v’=5 |
|
0 |
0.7294 |
0.2442 |
0.02715 |
0.001153 |
1.138E-5 |
1.028E-6 |
|
1 |
0.2155 |
0.3452 |
0.3641 |
0.0711 |
0.003072 |
1.055E-5 |
|
2 |
0.04553 |
0.286 |
0.1383 |
0.4238 |
0.1011 |
0.0045 |
|
3 |
0.007771 |
0.0932 |
0.2607 |
0.04305 |
0.4468 |
0.1433 |
|
4 |
0.001263 |
0.02393 |
0.144 |
0.2122 |
0.009505 |
0.4317 |
|
5 |
1.896E-4 |
0.005776 |
0.04983 |
0.1596 |
0.1566 |
8.132E-4 |
|
6 |
3.731E-5 |
0.001328 |
0.01248 |
0.06215 |
0.1581 |
0.1107 |
|
7 |
2.256E-5 |
3.281E-4 |
0.002952 |
0.01955 |
0.08192 |
0.1584 |
|
8 |
1.554E-5 |
5.921E-5 |
6.743E-4 |
0.005536 |
0.03042 |
0.09407 |
|
9 |
3.005E-6 |
2.328E-6 |
1.385E-4 |
0.001503 |
0.009449 |
0.03833 |
|
10 |
9.642E-7 |
1.158E-6 |
2.147E-5 |
3.58E-4 |
0.002644 |
0.01308 |
|
11 |
6.308E-6 |
1.02E-6 |
2.378E-6 |
5.804E-5 |
6.153E-4 |
0.00387 |
|
12 |
4.256E-6 |
8.552E-9 |
3.926E-7 |
4.986E-6 |
1.098E-4 |
9.261E-4 |
Ejercicio 3:
Tomar como intensidad
experimental de las bandas la altura de la cabeza de banda de
la Figura 3. Comparar la intensidad experimental con
la intensidad teórica obtenida mediante la ecuación (5) para
las bandas vibracionales indicadas en la Figura 3
(banda electrónica
d3Πg![]() a3Πu
de la molécula de
C2). Para el cálculo de la población del
estado excitado (ecuación 7) tomar para la temperatura de la
llama T=2000 K y normalizar, las intensidades teórica
y experimental, a la unidad para la banda más intensa
(
a3Πu
de la molécula de
C2). Para el cálculo de la población del
estado excitado (ecuación 7) tomar para la temperatura de la
llama T=2000 K y normalizar, las intensidades teórica
y experimental, a la unidad para la banda más intensa
(![]() ’=0-
’=0-![]() ”=0).
”=0).
Las curvas de energía potencial se representan en la Figura
4 (Hüber K.P y Herzberg G., Constants of Diatomic
Molecules. Van Nostrand-Reinhold. New York. 1979). De acuerdo con
la regla de selección ΔΛ=0,±1, las transiciones
A2Δ![]() X2Π y
B2Σ -
X2Π y
B2Σ -![]() X2Π están permitidas, tal y como se
observa en el espectro experimental (Figuras 1 y 2). El
espectro de la banda vibracional
X2Π están permitidas, tal y como se
observa en el espectro experimental (Figuras 1 y 2). El
espectro de la banda vibracional ![]() ’=0-
’=0-![]() ”=0 correspondiente a la transición
electrónica: B2Σ
-
”=0 correspondiente a la transición
electrónica: B2Σ
-![]() X2Π de la molécula de CH se muestra en
las dos figuras de la introducción y en la Figura 7, en
la zona entorno a 380-400 nm (25900-24800
cm-1). Al tratarse de una molécula con una masa
reducida pequeña (hidruro) las constantes rotacionales tienen
valores grandes (ver Tabla 3) con lo que la estructura
rotacional se puede resolver. El estudio detallado de la
estructura rotacional de una banda 2Σ
-
X2Π de la molécula de CH se muestra en
las dos figuras de la introducción y en la Figura 7, en
la zona entorno a 380-400 nm (25900-24800
cm-1). Al tratarse de una molécula con una masa
reducida pequeña (hidruro) las constantes rotacionales tienen
valores grandes (ver Tabla 3) con lo que la estructura
rotacional se puede resolver. El estudio detallado de la
estructura rotacional de una banda 2Σ
-![]() 2Π no será considerado aquí (para esta banda
electrónica, los estados corresponden al caso de acoplamiento
(b) de Hund; para cada valor de N hay dos niveles de
energía casi degenerados con J=N±½). Sin considerar el
espín electrónico (J=N), la energía rotacional se puede
expresar como (despreciando el término de distorsión
centrífuga y términos de orden superior)
2Π no será considerado aquí (para esta banda
electrónica, los estados corresponden al caso de acoplamiento
(b) de Hund; para cada valor de N hay dos niveles de
energía casi degenerados con J=N±½). Sin considerar el
espín electrónico (J=N), la energía rotacional se puede
expresar como (despreciando el término de distorsión
centrífuga y términos de orden superior)
![]() donde
donde ![]() (8)
(8)
siendo Be(=h/8π2cμRe2) , αe las constantes rotacionales en cm-1. Para una banda electrónica: Σ↔Π, la regla de selección entre estados rotacionales es: ΔJ=J’-J”=0, ± 1. Así tenemos 3 ramas; la rama Q (ΔJ=0), cuyas líneas vienen dadas por
![]() ;
J=J”=1, 2, 3, ...
(9)
;
J=J”=1, 2, 3, ...
(9)
donde ν0=Te’- Te”+G’(v’)-G”(v”) es el origen de banda (línea inexistente) y Bv’ y Bv” son las constantes rotacionales de los estados excitado y fundamental, respectivamente. Los números de onda de las líneas de la rama R (ΔJ=+1) vienen dados por
![]() ;
J=J”=0,1,2,... (10)
;
J=J”=0,1,2,... (10)
Los números de onda de las líneas de la rama P (ΔJ=-1) vienen dados por
![]() ;
J=J”=1,
2, 3,... (11)
;
J=J”=1,
2, 3,... (11)
Las primeras líneas de la rama P son P(1), P(2), P(3), ... donde se sigue la notación: RPQ(J”).
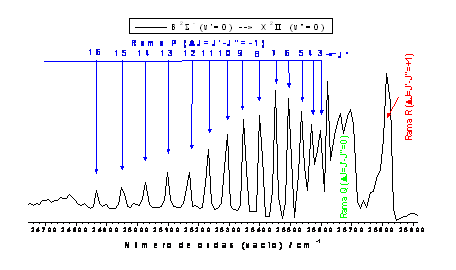
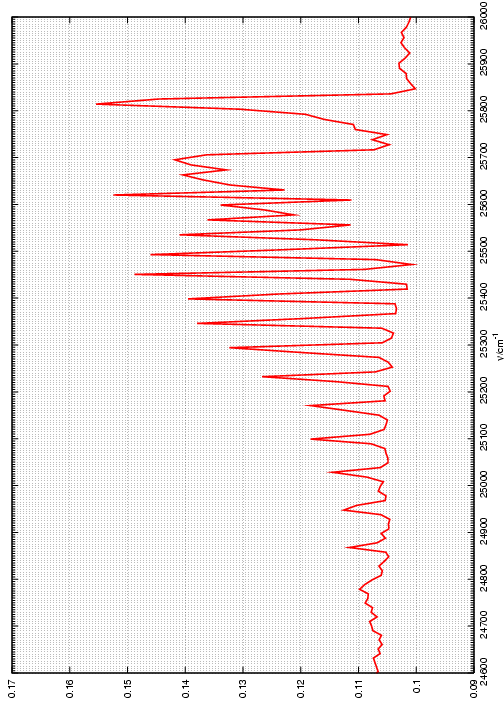
Figura 7: Emisión debida a la transición B2Σ --> X2Πde la molécula de CH y asignación de algunas líneas de la rama P.
Como vemos en la Figura 7, la banda está degradada hacia el rojo, formándose la cabeza de banda con las líneas de la rama R. El espaciado entre líneas consecutivas de la rama P viene dado por:
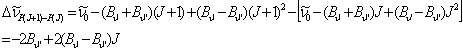 J=J”=1, 2, 3,...
(13)
J=J”=1, 2, 3,...
(13)
Ejercicio 4: A partir del espaciado medido de las líneas de la rama P indicadas en la Figura 7; Representar dichas diferencias en función de J=J” y del ajuste por mínimos cuadrados a la ecuación (13), determinar las constantes rotacionales Bv”=0 y Bv’=0. Comparar con los valores obtenidos mediante la ecuación (9) empleando las constantes de la Tabla 3.
(Pincha aqui si quieres la tabla con los puntos y aqui si quieres la figura en pdf, en postscript o en gif)Teniendo en cuenta que el número de moléculas NJ en un estado rotacional J, en un sistema en equilibrio térmico a una temperatura T, se puede tomar como
![]() (14)
(14)
es fácil obtener el nivel rotacional más poblado, que corresponde con el máximo [(dNJ/dJ)=0], obteniéndose
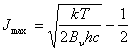 (15)
(15)
donde T es la temperatura absoluta en K y Bv es la constante rotacional en cm-1. A partir del valor medido experimental para Jmax (Figura 7), y del valor de B0, para la banda v’=0-v”=0, B2Σ - ->X2Π del CH; Determinar la temperatura de la llama empleando la ecuación 15.
Tabla 3:Contantes espectroscópicas experimentales para las moléculas consideradas (Hüber K.P y Herzberg G., Constants of Diatomic Molecules. Van Nostrand-Reinhold. New York. 1979.)
|
Molécula |
Estado Electrónico |
Te cm-1 |
cm-1 |
cm-1 |
cm-1 |
cm-1 |
Be cm-1 |
αe cm-1 |
γe cm-1 |
|
16O 1H |
X2Π |
0 |
3737.761 |
84.8813 |
-1.7915 |
0.32362 |
18.9108 |
0.7242 |
0.00706 |
|
OH |
A2Σ+ |
32684.1 |
3178.86 |
92.917 |
0.5409 |
-0.02134 |
17.358 |
0.7868 |
-0.016 |
|
12C 1H |
X2Π |
0 |
2858.5 |
63.02 |
|
|
14.457 |
0.534 |
|
|
CH |
A2Δ |
23189.8 |
2930.7 |
96.65 |
|
|
14.934 |
0.697 |
|
|
CH |
B2Σ- |
26070 |
2251 |
231 |
|
|
13.3815 |
1.4827 |
|
|
12C 2 |
a3Πu |
716.2 |
1641.35 |
11.67 |
|
|
1.6324 |
0.01661 |
|
|
C2 |
d3Πg |
20022.5 |
1788.22 |
16.440 |
0.5067 |
|
1.7527 |
0.01608 |
|
{kind=link}
{kind=link}